第二节 β-淀粉样肽
老年斑(senile plaques,SP)是AD患者大脑的主要病理特征。成熟的老年斑的核心为淀粉样物质,外面包绕成分有变性的轴索、树突,类淀粉样纤维、胶质细胞和突起。而SP淀粉样核心含有β-淀粉样肽(β-Amyloid peptide,Aβ)。
某些其他类型痴呆大脑也能出现类似的病理性淀粉样沉积,因此对Aβ正常、异常代谢的研究,可能对AD和多种痴呆脑病理发生、发展的认识及治疗有重要价值。
β-淀粉样肽为含40~43个氨基酸的多肽,是淀粉样肽前体蛋白(β-Amyloid precursor protein,β-APP、APP)的降解产物。中枢神经系统内所有细胞都能表达APP。Aβ的疏水残基位于C末端,说明C端位于质膜的磷脂双层,而N端位于细胞外。Aβ的C端最后几个氨基酸疏水性都很强,所以Aβ的C端越长,沉积性越强,神经毒性越大。脑内产生的Aβ主要形式是Aβ40,部分为Aβ42和Aβ43。细胞水平研究显示,用Aβ寡聚体处理体外培养的神经元显示有毒性作用,能引起胶质细胞炎症反应(图5-2-1)。
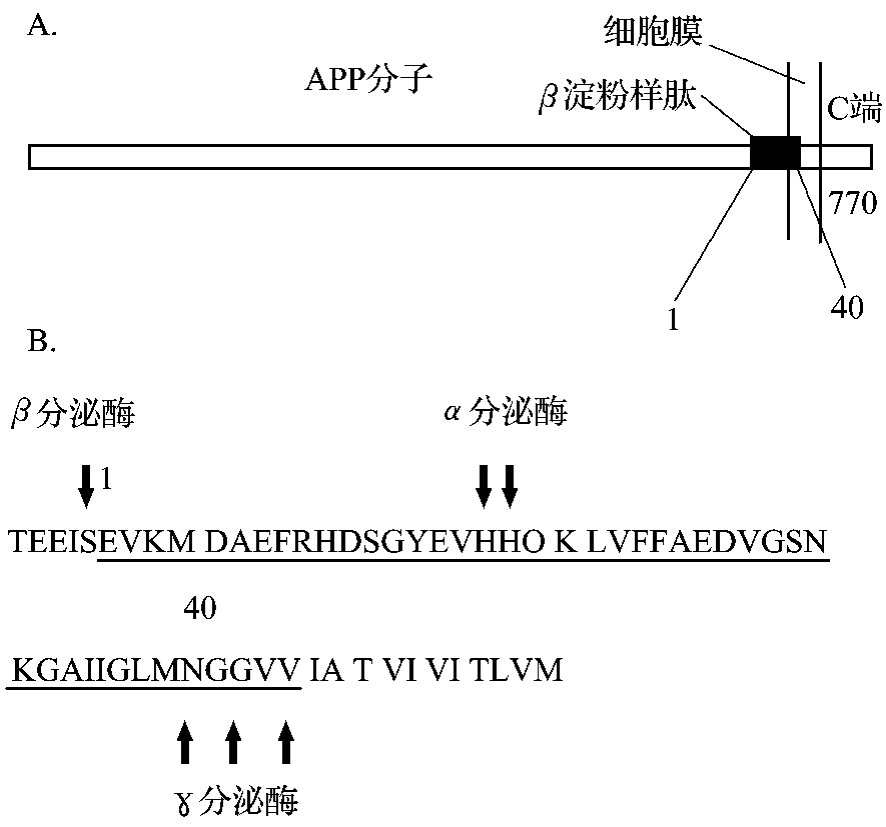
图5-2-1 Aβ的氨基酸序列和在APP分子中的部位。A.Αβ在APP分子中的部位;B.Αβ的氨基酸序列及在前体蛋白中各种分泌酶的裂解位点
正常人的脑脊液也含有Aβ,但正常人Aβ不在大脑中沉积。说明正常APP加工产生可溶性Aβ。而AD患者大脑的老年斑和血管壁上都可见Aβ沉积。人们认为,老年斑中主要淀粉样成分是Aβ42和Aβ43,AD脑血管壁的类淀粉物质多为Aβ40。Aβ脑内沉积可能发生于AD病理的早期,Aβ可能作为生长趋向因子,再诱导变性轴索、星形胶质和小胶质细胞围绕Aβ蛋白形成老年斑块。
Aβ由前体蛋白APP通过分泌酶类水解加工生成,部分经细胞内中性内肽酶等降解清除,Aβ水平决定于产生和分解的动态平衡。因基因缺陷可能直接或间接增加APP表达或改变相应蛋白酶分解过程,可能影响Aβ聚集和稳定性,引起AD病理综合征。这些过程改变了Aβ产生和清除间的动态平衡,造成的聚集态的Aβ累积又诱发下游一系列复杂病理反应,包括突触/突起的变化,tau蛋白过度磷酸化,神经递质损失,神经胶质增生和炎症反应加强,最后引起神经元功能障碍、凋亡,表现出老年斑块和神经原纤维缠结等典型病理特征。
一、Aβ沉积是AD病理发生的诱因
近乎所有AD患者的大脑都有大量含Aβ淀粉样斑块沉积和神经末梢退化等病理改变,在海马等相关学习记忆脑区的斑块数量与脑力损害程度正相关。对家族性AD的遗传学研究发现,4个AD致病相关基因都与Aβ代谢、累积相关。其中APP、早老蛋白1和2(PS1,PS2)基因的突变都能引起Aβ生成增加[5]。
关于PS的生理作用,一方面,PS1和PS2主要存在于细胞内质网内,小部分存在于高尔基体内,而APP主要集中于高尔基体,有利于PS在细胞器内通过与APP以非共价键相互作用,调节APP的加工、运输和蛋白水解等过程。如PS2过表达可使APP分泌片段减少。另一方面,PS自身是Aβ生成关键酶γ-分泌酶活性的必需组分。
ApoE4基因是AD的一种风险基因,ApoE4基因频率高的个体增加患AD风险。而ApoE4可在AD临床症状发生前诱导Aβ的过量积聚,影响Aβ沉积。
APP为生成Aβ的前体分子,过量APP表达会引起Aβ累积增加。只要基因遗传、变异能引起少量APP表达增加,都有导致迟发性AD疾病的危险。在第21对染色体上存在APP基因位点的重复,如患者有21染色体三倍体的染色体畸变可患唐氏综合征。由于APP基因过量表达使该病患者Aβ分泌早年就增加,十几岁出现Aβ斑块沉积,较晚会出现神经纤维缠结等其他生化病理改变。患者最终都表现早老性痴呆症状。已经证明,单纯增加APP基因的拷贝数而没有APP基因突变也可导致AD早期发病。患者大脑AD发病进程伴随大量淀粉样沉积物和神经纤维缠结等典型病理特征。
如果有方法能减缓病源性淀粉蛋白产生,就有望用于人类的AD等淀粉样神经变性疾病的有效治疗。阐明Aβ沉积的发生发展机制,不但可能发现临床治疗AD等神经变性疾病的方法,还有利于人类学习记忆功能机制的破解。
二、Aβ神经毒性和AD病理发生
各种病因诱发的AD中,Aβ累积和沉积是共有病理过程。Aβ累积和沉积是AD病理发生和发展的始动因素和关键事件,其机制涉及以下方面。
Aβ可能诱导神经细胞凋亡。第一,Aβ的过量产生,APP非淀粉样代谢途径明显减少,使神经营养成分sAPPα生成减少,增加神经细胞对多种损伤的敏感性。第二,某些突变的APP和PS1、PS2基因产物可结合其他蛋白引起细胞死亡。Aβ的聚集状态和构象决定其神经毒性。经Aβ处理24 h的神经元,表现凋亡的特征性生化改变,如DNA带呈现梯状分布。Aβ慢性处理的神经元,能显示轴突营养不良,染色体固缩等细胞凋亡形态学特征。Aβ毒性能影响凋亡相关蛋白表达,如下调细胞抗凋亡基因Bcl-2的表达。
Aβ通过激活胶质细胞诱发中枢神经系统的炎性反应。AD病理常伴有慢性炎性病理过程。研究证实,Aβ可持续激活炎症修复机制,使正常急性反应转变为慢性炎症损伤。在Aβ沉积部位可见小胶质细胞过度激活和白介素1(IL-1)表达。神经元周围沉积的Aβ与小胶质细胞膜CD36和CD47等受体复合物相互作用,促进小胶质细胞活化、增殖,分泌大量促炎因子,引起炎症损伤。IL-1过度表达和释放作为始动环节,增加胶质细胞的其他炎性细胞因子表达和各种胞外炎症分子的生成。这使得在AD病理损伤的全过程都有慢性炎症产物发挥作用[6]。
Aβ神经毒性可通过多种途径导致氧化应激,进而导致脑神经细胞损伤、死亡。大脑代谢耗氧量大,不饱和脂肪酸含量多,抗氧化酶活性不足,最易受自由基损伤。自由基损伤可能是Aβ的主要毒性机制。
Aβ自身是氧自由基供体,可产生活性氧,还可诱导氧自由基产生,如超氧阴离子、羟自由基等。Aβ激活小胶质细胞,也促进大量自由基生成,加剧氧化应激。实验表明Aβ沉积与氧自由基的产生直接相关,Aβ沉积的斑块聚集物内生成大量氧自由基。AD患者脑表现过度的蛋白质氧化和脂质过氧化,抗氧化酶活性降低。研究证明,维生素E等抗氧化剂能阻断Aβ的神经毒性作用。
Aβ可能结合神经细胞膜表面受体,跨脂质双层膜形成Ca2+通道,引发Ca2+内流致胞内Ca2+超载,降低神经元膜流动性。Aβ可增加第二信使IP3含量,作用内质网的IP3受体,引起Ca2+释放,增加胞内Ca2+。细胞内钙稳态的失衡,进而造成细胞膜的完整性破坏,增加兴奋性递质释放,引起神经元损伤。膜流动性降低可降低膜酶活性、膜受体功能及影响神经信号传递相关过程。
三、Aβ细胞分布、分子形式与神经毒性
现在认为,Aβ存在于AD斑块中,但AD疾病进程后期才形成Aβ沉积斑块,斑块不是诱发AD的原因。小分子、可溶性Aβ寡聚体可能是诱发疾病的早期原因,但具体机制仍不清楚。
对Aβ毒性的细胞分布,现在研究提示细胞内Aβ可能是AD发病的早期标志。很多研究认为,Aβ产生后立刻被分泌到胞外起作用,近来通过转基因鼠的研究发现,细胞内Aβ聚集早于细胞外,细胞内外的Aβ性质有一定差异。在神经细胞膜两侧可溶性Aβ可能因内外环境不同形成不同结构的聚合体。而细胞内Aβ可能是起始神经元退行性病变和斑块形成的决定因素。美国LaFerla实验室用三联转基因小鼠(APP,TAU及PS1)研究神经元内Aβ聚合的过程,4月龄时神经元内出现明显可测的可溶性、非聚合态的Aβ;6月龄时海马等脑区可见寡聚体形成;1年后,Aβ寡聚物从细胞内转移到胞外,多见于斑块附近。细胞内寡聚物存在于近胞体和近突触的轴突部位[7]。在神经元内寡聚物可与tau蛋白共存,但在老年鼠却不与高磷酸化tau共存。APP/PS1转基因鼠中细胞内Aβ的积累也早于神经元死亡。
AD大脑表现Aβ在细胞外产生聚集纠结和沉积。还有高度磷酸化tau蛋白聚集形成的细胞内神经原纤维缠结(NFT)。tau蛋白磷酸化后丧失其稳定微管的能力,影响细胞存活。但部分AD患者大脑中,NFT存在不明显。NFT在许多神经疾病中存在,可能是一种非特异的神经损伤性病理过程。
Aβ诱发AD病理发生和发展的神经毒性主要涉及下列几方面。(https://www.daowen.com)
(一)Aβ影响神经元突触功能
用培养转基因鼠神经元研究证明,激活突触活动使细胞内Aβ水平下降,Aβ释放增多。检查表明,在AD发病过程中神经元输出逐渐降低,这可能使AD疾病过程中Aβ释放降低,神经元内Aβ累积增加,加剧神经元的退行性病变;也可能是AD病理过程中Aβ生成减少,使Aβ释放相应降低,这些关键问题还需要研究解决。
美国研究者报道,神经细胞Aβ水平增加能影响受体蛋白的运输。如增加Aβ的释放可能通过改变谷氨酸Ⅱ型受体的运输,抑制AMPA及NMDA受体介导的海马突触神经传导。Aβ可增加网格蛋白小窝介导的谷氨酸Ⅱ型受体的内吞作用,抑制突触传递。研究还证明,除谷氨酸Ⅱ型受体外,Tg2576转基因小鼠中谷氨酸Ⅰ型受体也可下调。有人发现转基因小鼠神经元表面谷氨酸Ⅰ型受体表达下调,同时一种使GluR1锚定在突触的蛋白PSD-95表达同步下调,PSD-95是膜性的鸟苷酸激酶,与谷氨酸受体回收及锚定突触后密度相关。可能涉及Aβ影响受体运输的机制,
Aβ也可能通过调节内涵体输送通路影响细胞表面受体的分布和作用。晚期的内涵体递送膜蛋白至溶酶体降解,Aβ42与晚期的内涵体共存。Aβ可能通过调控泛素蛋白酶体系统(UPS)影响内涵体的回收。APP转基因动物表现蛋白酶功能受损,阻断受体及其他突触元件回收循环过程。
有人研究电刺激过程中海马间质液Aβ(ISF Aβ)数量的变化,发现电刺激清醒的自由活动小鼠,海马间质液Aβ水平明显上升,用河豚毒素阻断正常的神经活动,ISF Aβ下降。特异药物阻断神经递质小囊泡的释放,8 h内ISF Aβ水平下降80%。说明虽然Aβ不通过突触囊泡转运,但突触囊泡的回收过程可能影响Aβ在突触的活动。
上述结果提示,突触的活动能影响Aβ在细胞内外的传递,而细胞内外Aβ寡聚体的量也影响着突触相关受体的运输。
(二)Aβ影响重要信号通路
有人证明,细胞内Aβ干扰神经元保护性的信号通路和细胞的应激反应。丝/苏氨酸蛋白激酶B(Akt)在大脑中起着重要作用,在多种大脑疾病中都表现Akt异常。原代培养的神经元中Akt激酶过表达,可增加其对细胞内Aβ毒性累积的抵抗力,有保护作用。反之,Aβ处理原代培养的皮层神经元可降低细胞内Akt的磷酸化激活,而活化的Akt可以抑制以tau为靶点的激酶GSK3β,Akt受阻导致GSK3β激活。动物水平研究有类似结论[8]。
在AD病理早期,细胞内Aβ的毒性可能通过阻断Akt的激活,减弱细胞的应激反应,表现异常的细胞信号通路。某些神经保护剂如VEGF、BDNF和IGF-1等是以激活Akt信号途径为靶点,其效用受到关注。
细胞膜的两侧Aβ存在着动态平衡。细胞外Aβ可有神经毒性。Aβ寡聚体加到培养细胞外时,Aβ可能通过细胞的内吞作用进入细胞。研究者将正常小鼠脑组织移植至转基因小鼠脑内,发现3个月后在移植物及其他脑区有明显斑块形成,且在移植区斑块形成更快更多,Aβ斑块附近神经元有轻微的病理损害。证明细胞外可溶性的Aβ能够扩散进入正常的神经组织再形成斑块,并表现出一定的病理变化。细胞外的Aβ能够诱导细胞的病理生化改变。
细胞内和细胞外的Aβ似乎都影响突触功能。转基因鼠脑神经元Aβ生成增多与突触改变明显相关,而全长APP水平并未升高。以Aβ生成关键酶β-分泌酶(BACE-1)抑制剂阻断Aβ生成,能逆转该转基因小鼠认知功能损害症状。这说明引起AD的原因是Aβ的积聚而不是APP过多。以上述PSD-95和GluR1的改变作为影响突触功能的指标,证实细胞外Aβ也能影响突触改变,Aβ引起的突触改变是可逆的。扩散性Aβ寡聚体或较大的Aβ聚合物可能在体内都能引起神经毒性,但由于Aβ寡聚体是早期的病理改变,同时其毒性作用是可逆的,所以是较好的治疗靶点。
(三)Aβ寡聚体形态和神经毒性
现在认为,Aβ聚集状态影响Aβ毒性。有损害神经元功能作用的Aβ是一类小分子寡聚体。不同Aβ聚集态表现不同毒性,因此,老年脑内有弥散性斑块有时并不引起痴呆症状。
研究者利用合成分子制备成稳定的12Aβ分子Aβ42寡聚体,分子量60 kD。研究证明,AD患者和Tg2576转基因鼠的大脑皮层存在可溶性的这种Aβ聚体,正常人脑脊液中不存在。该Aβ42寡聚体能完全阻滞离体脑片的长时程增强效应(LTP)[9]。
合成Aβ经体外孵育可产生的大小为4、8、16、18 kD的可溶性聚合物,称为扩散性Aβ寡聚体(ADDLs)。ADDLs可能通过形成更大的聚合体(48~56 kD)形态,在体外能诱导细胞凋亡,抑制神经细胞存活。对脑片诱导的LTP的抑制表明ADDLs自身有突触毒性。ADDL作用于原代培养的海马神经元数小时后,能引起磷酸化tau蛋白水平增加并激活磷酸化激酶GSK3β。动物脑室注射Aβ寡聚体能激活GSK3β,表现认知障碍。Aβ寡聚体可能作为淀粉样肽原发毒性形式。具神经毒性的Aβ寡聚体形成和作用,可能是导致AD病理形成,突触功能损伤的关键。因此,研究者重视探索Aβ在体内聚集为Aβ寡聚体,原纤维或形成纤维斑块的过程和抑制方法。
四、抑制Aβ的聚集和沉积的AD治疗策略。
(一)抑制Aβ的聚集
Aβ的聚集与其β片层结构密切相关。在溶液中,Aβ42倾向于形成斑块样的沉积,而Aβ40则更易于形成典型的纤维。研究证明,星形胶质细胞能直接降解Aβ。因此,加强星形胶质细胞对Aβ的清除作用能降低Aβ的神经毒性,缓解AD的症状。
研究发现,由五个氨基酸组成β片层阻断肽(iAβ5p)能抑制Aβ的β片层结构阻止Aβ纤维形成,且能分解Aβ纤维,对神经元有保护作用。注射动物后1个月后可部分减少已形成的Aβ沉积,注射部位星形胶质细胞明显减少。可能成为治疗AD的一种方法。另外,β环糊精、烟碱也有抑制Aβ单体的聚集、Aβ纤维形成,减少Aβ诱导的神经毒性等作用。
(二)促进Aβ的降解和清除
正常脑内Aβ产生和清除是一个动态平衡过程。中性内肽酶(neprilysin,NEP)是金属蛋白酶家族的一员,能降解脑内部分Aβ。药物上调脑内NEP的表达可能减少或预防SP形成。转染人NEP基因表达载体后AD模型鼠脑内Aβ沉积和已存在的斑块都减少。神经退行性变和细胞损害减轻[10]。胰岛素降解酶(insulin dedgrading enzyme,IDE)和内皮素转化酶在体内也可降解AD病理过程中的Aβ。
(三)抗Aβ免疫治疗
Aβ免疫治疗分主动免疫和被动免疫。主动免疫应用Aβ多肽疫苗;被动免疫应用抗Aβ的抗体。在Tg2576等转基因小鼠给予Aβ抗体,能促进清除Aβ沉积,缓解认知功能下降。AD小鼠经外周注射Aβ抗体后,抗体可进入脑内,黏附于Aβ沉积物并促进周围小胶质细胞激活、吞噬Aβ沉积。体外Aβ抗体处理小胶质细胞共同培养的Aβ沉积物,能激活小胶质细胞,明显减少Aβ沉积。
临床Ⅱ期实验显示,Aβ疫苗能使认知稳定,缓解痴呆进程。神经病理学检查发现,可使脑内Aβ减少,但不能改变AD相关病理特征现象。问题在于,约有5%的Aβ疫苗受试患者产生了脑膜脑炎。因此,Aβ免疫治疗的疗效和安全性还需要更多实验和研究。
(崔 行)
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。





