金属型氢化物主要是指氢元素与电负性低而化学活性大的IA、IIA族等元素结合形成的诸如LiH、MgH2等离子键化合物,与过渡金属和类过渡金属反应形成的诸如TiH2等过渡型氢化物,以及储氢合金吸氢后的氢化物。多元络合氢化物,又称为配位氢化物,是典型的多元氢化物,如NaAlH4、Mg(BH4)2、LiNH2等。这两类储氢材料由于其优越的性能而吸引很多学者围绕其展开了一系列的对应计算。
1.金属型氢化物
金属型氢化物的吸放氢反应可以概括表示为

式中,M为某种金属或金属化合物;MHx为反应产物即氢化物;ΔH为反应焓变。
较早地,人们运用从头算法和密度泛函理论(DFT)等方法对LaNi5、LaNi4Al、La2Ni4Mn等体系的化学键、生成焓[3]、电子结构以及成键特征[4]等方面进行了研究。Tatsumi等人[5]研究了LaNi5-H固溶体的原子结构和热力学性质,计算得出12n位的能量最低,其次为6m位;Hector等人[6]比较了LaNi5H3和LaNi5H7的计算结果,得知H原子首先排布在晶体中上层的间隙,并且优先占据12n和6m位。但对镧基储氢材料吸/放氢的本质、吸/放氢的机理以及镧基储氢材料与氢气相互作用研究很少。
镁基储氢是之后的又一个研究热点。Mg2Ni是六方晶系,吸氢反应焓变为-64.48kJ/mol H2生成Mg2NiH4,高温氢化比低温氢化容易且动力学性质好,在523K下,该氢化物为晶格常数a=0.6507nm的立方HT相(空间群Fm2m),温度降低时Mg2NiH4转化为复杂的单斜LT结构(空间群C2m)。晶体结构如图11-4所示。
MgH2属离子型氢化物,含氢量为7.65wt%,ΔHΘ=-74.48kJ/mol H2,放氢温度过高,吸放氢速率较慢,因此人们试图采用计算的方法来解释和预测改善的方法。Vegge[7]采用DFT和速率理论研究了氢分子在Mg(0001)面的解离和随后氢原子扩散到镁基底,结果显示氢气在镁(0001)面桥位上的解离能垒高达1.15eV,氢原子从表面向亚表层扩散并最终向体相扩散的能垒分别为0.53eV和0.18eV,所以Mg储氢材料吸氢过程的速控步骤为氢气的解离。Jiang等人[8]发现氢气低密度吸附在Mg(0001)表面时倾向于表面的面心位点,直到达到单层全部吸附时结构能量才稳定,另外还分析了MgH2形成过程。Krijn P.de Jong等人[9]以MgH2为研究对象,基于DFT的第一性原理计算发现在一定的纳米尺度范围内,储氢材料的脱附焓随着材料尺寸的减小而减小,当粒径小于1.3nm时,MgH2纳米颗粒的脱附焓将会随粒径的减小而发生显著的变化,因而可以通过控制材料的尺寸来达到理想的储氢性能。Grossman等人[10]利用量化蒙特卡洛(QMC)方法研究MgH2体系的纳米团簇,对比运用其他不同交换相关泛函的DFT结果,发现高精度的DFT:DMC的方法对于金属氢化物纳米效应的模拟更合适。
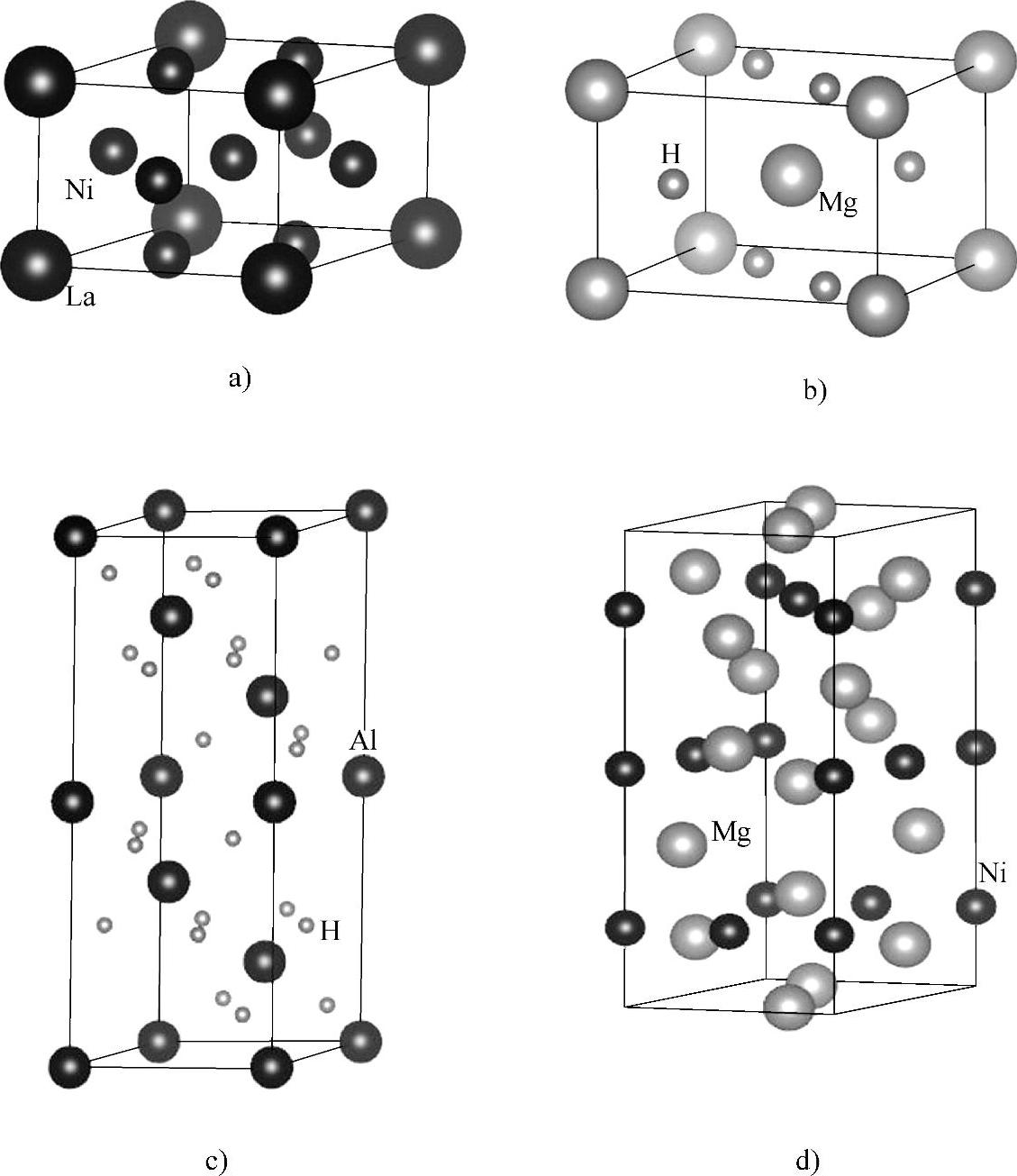
图11-4 晶体结构示意图
a)LaNi5 b)MgH2 c)AlH3 d)Mg2Ni
金属氢化物的晶体结构对其吸放氢有较大影响,是理论储氢容量计算的依据,而过渡金属氢化物因其结构相似于MgH2,所以也备受关注。Tao等人[11]计算了Mg和Ti以吸收的H原子浓度作为自变量的吸附能,结果发现Mg和Ti的氢化物最稳定结构分别是金红石和萤石结构,在Mg中氢原子偏于结对形成团簇,而Ti中单独占位并互相远离。随后Tao又针对MgH2和过渡金属氢化物TMH2插层设计建模,预测是一种潜在的有吸引人的热力学性质的新型储氢材料。
文献报道AlH3有8种物相,大部分都没有指标化或得到晶体结构,其中稳定的α-AlH3空间群R-32/c(167),拥有六方对称性[12]。Ke等人[13]计算并预测了立方和斜方相的另外两种稳定的AlH3,探明了晶体结构、电子结构和热力学性质等,发现和α-AlH3一样有负的形成焓和正的形成自由能,与Wolverton等人[14]的结论一致。
在金属氢化物的实际应用中,反应速率是需要重点考虑的因素,即探讨氢化/脱氢反应规律、考虑金属氢化物氢化/脱氢反应动力学模型。研究发现,金属氢化物体系的反应动力学研究既要在气固相反应一般理论的指导下进行,又要考虑该体系的特点进行针对性的处理[15]。
2.多元络合氢化物(https://www.daowen.com)
多元络合氢化物,又称为配位氢化物,是典型的多元氢化物,如NaAlH4、Mg(BH4)2、LiNH2等,形成这类氢化物的结构基础是IIIA族元素的氢化物BH3和AlH3,是缺电子的不稳定集团,它们与负氢离子结合成为四面体结构的络离子BH4-和AlH4-。
(1)Al/B系复杂氢化物
该系复杂氢化物化学通式Ax(MH4)y形式,A为碱金属或碱土金属,M主要是Al或者B。
NaAlH4通常呈α相,它是体心四方结构,空间群是第88号的I41/a。每个晶体学原胞含2个NaAlH4化学式单位。其中Na占据的Wyckoff位是4a(0,1/4,1/8);Al占据的Wyckoff位是4b(0,1/4,5/8);H则占据16f(0.2372,0.3836,0.5469)Wyckoff位。Al和最近邻4个H形成一个接近正四面体的AlH4基团。这种物质曾多被作为研究的样板[16-20]。
Yildirim小组[21-23]用理论实验相结合对Ti掺杂NaAlH4做了深入研究,对体系掺杂前后的结构和总能量进行了计算比较,得知Ti优先取代Na,这种取代掺杂弱化了Al-H键从而使得氢化物更易脱氢。Herbst等人[24]基于DFT对AGaH4(A=Li、Na、K、Rb和Cs)的7种基态晶体结构进行了系统计算,发现LiGaH4和NaGaH4的标准形成焓在引人注意的-30kl/mol H2左右。
对于硼氢化物,Setten等人对Mg(BH4)2计算结果显示室温下生产MgB和H2的反应焓变38kJ/mol H2[25],随后研究又猜测并证实了MgB2H6新相存在[26];Caputo等人[27]以第一性原理方法为主,联合多种计算方法,建立并计算出Mg(BH4)2的最小能量的基态结构,并进一步发现金属原子的配位是碱金属和碱土金属硼氢化物的晶体结构的主要影响因素;Lee等人[28]利用第一性原理和CALPHAD模型研究发现,Mg、Ca和Zn掺杂离子降低了LiBH4的稳定性,其中Mg和Zn对分解温度影响很弱,Ca掺杂因形成高稳态CaB6而明显降低H2的释放温度;Kim等人[29]针对LiK(BH4)2、KBH4和NaBH4晶体结构和热力学性质,Ti掺杂的LiBH4中H的相互作用也有相关密度泛函研究[30]。晶体结构如图11-5所示。
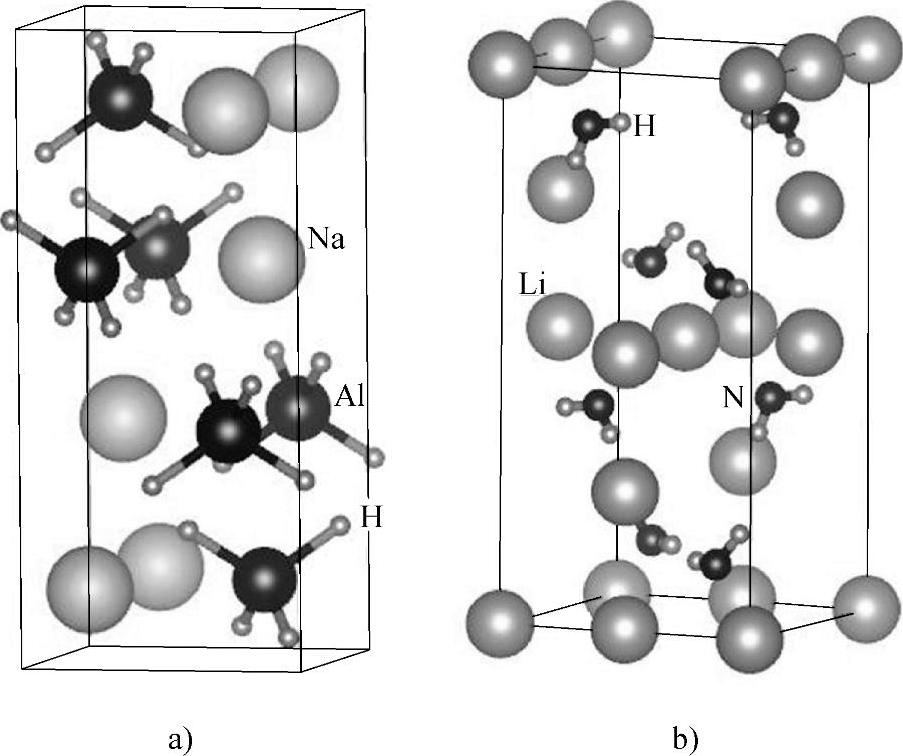
图11-5 晶体结构示意图
a)NaAlH4 b)LiNH2
(2)氨基和亚氨基化合物
氨基/亚氨基化合物也是储氢材料研究的热点之一。为了改善其储氢性能,很多针对电子结构、吸放氢反应的能量等基础性质的研究得以展开。
Wang等人[31]运用第一性原理计算了Li-N-H体系作为储氢材料的电子结构、化学键型和热稳定性,计算焓变和实验测得焓变大致吻合,分别为-75.67kJ/mol H2和-69.17kJ/mol H2,与前人数据基本一致。Seip等人[32]基于DFT研究了Mg(NH2)2体相、表面和团簇,确认该结构为l41/acd四方相,对比5个不同表面发现(112)表面最稳定,电子态密度计算结果对比说明团簇的能带占据态是纳米相有着增强的动力学的一个原因,另外发现脱去NH3比H2中能量上更易实现,事实上这是金属-N-H体系的一般趋势。Liu等人[33]计算了Li2 Mg(NH)2结构和电子性质,优化的晶胞参数和键长与实验数据一致,α型系基态构型,布局分析显示N-Li/Mg离子型而N-H作用弱于β型,价带受N的s和p轨道影响显著。Wang等人[34]研究了LiNH2、Mg(NH2)2和Li2MgN2H2在吸放氢反应中氨基离子的作用,Cui等人[35]研究了Li3N在高压下的结构转变。
(3)复合型氢化物
多元化的金属氢化物复合在一起在实验上得到推广,相关计算研究也随之开展。一般地,该类体系化合物由于晶体结构相对比较复杂,计算量大。Chater等人[36]合成并分析了Li4BH4(NH2)3的晶体结构;Akbarzadeh等人[37]对Li-Mg-Al-H体系研究深入;Farrell等人[38]第一性分子动力学研究了Li4BN3H10在熔点上下温度范围的结构和动力学行为。Ozolins等人利用PEGS与GCLP相结合的新方法,针对Li-Mg-N-H[39]、Li-Mg-Ca-B-H[40]、(NH4)2B12H12[41]体系做了一些出色的工作。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。






