VASP是Vienna Ab-initio Simulation Package的缩写,中文全称维也纳从头计算模拟包,采用平面波赝势(或缀加投影波)方法进行从头计算分子动力学模拟,是当今国际上应用最为广泛的从头计算的商业软件之一。该程序流程示意图如图11-3所示。目前该软件包最新版本5.2,网站地址为:http://www.cms.mpi.univie.ac.at/VASP。Mede-A VASP为其提供了友好界面。
VASP中的方法基于有限温度下的(对电子气而言)局域密度近似,自由能作为电子气密度的泛函在每个MD时间步长内精确求解电子气的瞬时基态。主要有以下亮点:采用了缀加投影波(PAW)或超软赝势(USPP),减少了平面波的数目[特别是对过渡金属和第一列的元素(C和O)],有完善的赝势库;在实空间中计算势的非局域部分并保持正交化的数目较少,使得计算时间小于N3,Nmax=4000;在电子的自洽迭代计算中,采用了非常有效的算法(如RMM-DISS和Blocked Davidson),可实现有效、稳定、快速的Kohn-Sham方程自洽求解;能自动确定体系的对称性并由此进行计算;几乎支持所有计算机平台。
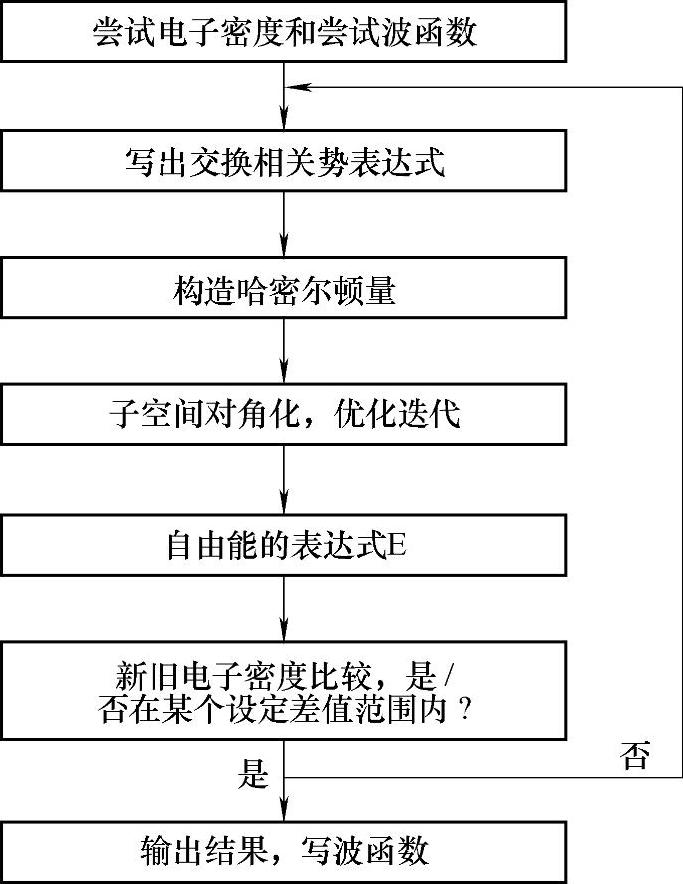
图11-3 程序流程示意图(https://www.daowen.com)
可实现的计算功能主要有:采用周期性边界条件(或超原胞模型)处理原子、分子、团簇、纳米线(或管)、薄膜、晶体、准晶和无定性材料,以及表面体系和固体;计算材料的结构参数(键长、键角、晶格常数、原子位置等)和构型;计算材料的电子结构(能级、电荷密度分布、能带、电子态密度);计算材料的状态方程和力学性质(体弹性模量和弹性常数);计算材料的光学、磁学和晶格动力学性质(声子谱等);计算材料的激发态(GW准粒子修正);表面体系的模拟(重构、表面态和STM模拟)以及从头分子动力学模拟等。
使用VASP进行计算,用户需要准备输入文件才可以进行。VASP的输入文件主要有4个,分别为INCAR(主要参数输入文件)、KPOINTS(K点取样设置文件)、POSCAR(描述体系结构的文件)和POTCAR(赝势文件)。输出文件类型较多,根据计算内容分析相应的输出文件即可。更多说明请查看VASP Manual和官方论坛。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。





