近年来,随着量子力学、凝聚态物理和计算技术等基础学科的发展,“计算模拟”已与实验、形式理论成三足鼎立之势,形成科学发现的第三条途径,在材料和化学领域的推动和贡献愈加明显。“实践是检验真理的唯一标准”是材料计算模拟发展历程中的基本思想,作为理论基础的第一原理具有公理结构,选用适当的物理模型,坚持“严”(机理正确)和“密”(数值准确)的科学思想,材料计算模拟与多学科交叉融合,目的在于理解、预言和发现新的物理化学现象及其科学本质,已发展到包括计算量子化学、材料计算与设计、分子模拟、计算化学和统计力学等领域。
储氢材料目前发展迅猛,前面章节已有分类具体介绍,但仍属相对基础科学的研究课题,要真正理解其中新的化学现象和概念,不断揭示和探索出新型储氢材料,计算模拟是不可或缺的可靠研究方法。材料计算模拟的大致流程如图11-1所示。大概说来,对储氢材料进行多尺度计算模拟,可以得到包括晶体结构和成键特征、电子结构、结合能、生成焓和脱附焓以及由此估算的脱氢温度等物理化学性质、量子力学性质以及统计力学性质等信息。
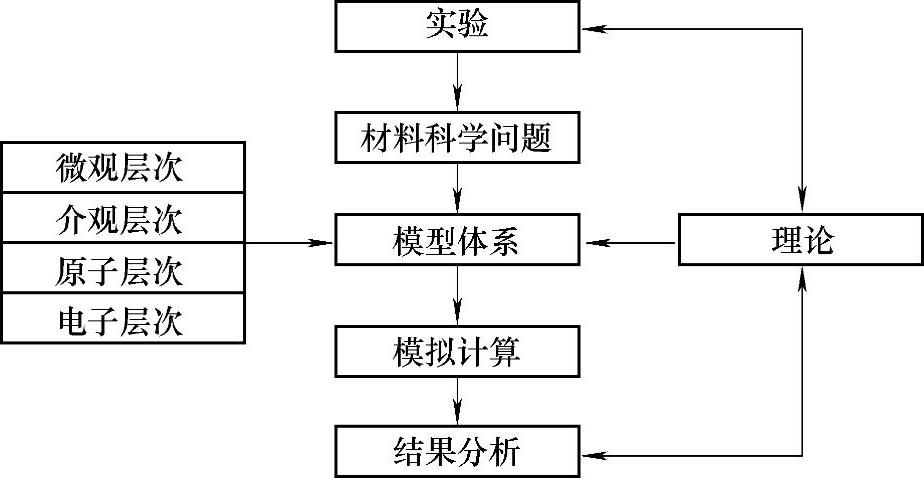
图11-1 材料计算模拟流程示意
结构决定性质,晶体结构优化是一切性质计算的基础。结构与能量紧密相关,一般定义使得体系具有最低能量的结构是最稳定的。能量的分析可以预测分子级别的过程能否进行。个体系的能量通常分解为动能和势能,其中动能可进一步分解为振动能、平动能与转动能,势能可由Coulomb定律描述,也可进一步分解为键伸缩势能、摇摆势能、形变能、氢键能等。固然不同的计算方法得到的绝对能量是不同的,但相对能量一般是准确的。(https://www.daowen.com)
体系的大多数性质都决定于它的电子结构,即电子的空间与能量分布。第一性原理计算多重视电荷密度分布、电子局域函数、电子态密度和能带结构等电子结构性质。
热力学性质定义了很多能量,诸如内能、自由能和熵等。计算得到结合能、生成焓和脱附焓以及由此估算的脱氢温度,有利于帮助理解储氢材料的真实物理化学性质。但从热力学的数据无法获得化学过程的详细描述,进一步还需要动力学的计算模拟。
量子力学是电子行为的精确数学描述,因此也是化学的精确数学描述。统计力学是从材料的分子描述来计算材料体相的热力学性质的数学手段,通常运用分子动力学或者Monte Carlo(蒙特卡洛)计算来得到统计数据,得到可能出现哪些构型及其出现概率,然后可以设置恒容、恒压或者恒温的模拟来联系体系热力学性质。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。





